




El Premi Lasker de Recerca Mèdica Bàsica d'enguany va ser atorgat a Demis Hassabis i John Jumper per les seves contribucions a la creació del sistema d'intel·ligència artificial AlphaFold que prediu l'estructura tridimensional de les proteïnes basant-se en la seqüència de primer ordre d'aminoàcids.
Els seus resultats resolen un problema que durant molt de temps ha preocupat la comunitat científica i obren la porta a l'acceleració de la recerca en tot el camp biomèdic. Les proteïnes tenen un paper fonamental en el desenvolupament de malalties: en la malaltia d'Alzheimer, es pleguen i s'agrupen; en el càncer, la seva funció reguladora es perd; en els trastorns metabòlics congènits, són disfuncionals; en la fibrosi quística, van a l'espai equivocat de la cèl·lula. Aquests són només alguns dels molts mecanismes que causen malalties. Els models detallats de l'estructura de proteïnes poden proporcionar configuracions atòmiques, impulsar el disseny o la selecció de molècules d'alta afinitat i accelerar el descobriment de fàrmacs.
Les estructures de les proteïnes es determinen generalment mitjançant cristal·lografia de raigs X, ressonància magnètica nuclear i criomicroscòpia electrònica. Aquests mètodes són cars i requereixen molt de temps. Això fa que existeixin bases de dades d'estructures de proteïnes en 3D amb només unes 200.000 dades estructurals, mentre que la tecnologia de seqüenciació d'ADN ha produït més de 8 milions de seqüències de proteïnes. A la dècada de 1960, Anfinsen et al. van descobrir que la seqüència unidimensional d'aminoàcids es pot plegar espontàniament i repetidament en una conformació tridimensional funcional (Figura 1A), i que les "xaperones" moleculars poden accelerar i facilitar aquest procés. Aquestes observacions condueixen a un repte de 60 anys en biologia molecular: predir l'estructura tridimensional de les proteïnes a partir de la seqüència unidimensional d'aminoàcids. Amb l'èxit del Projecte Genoma Humà, la nostra capacitat per obtenir seqüències d'aminoàcids unidimensionals ha millorat molt, i aquest repte s'ha tornat encara més urgent.
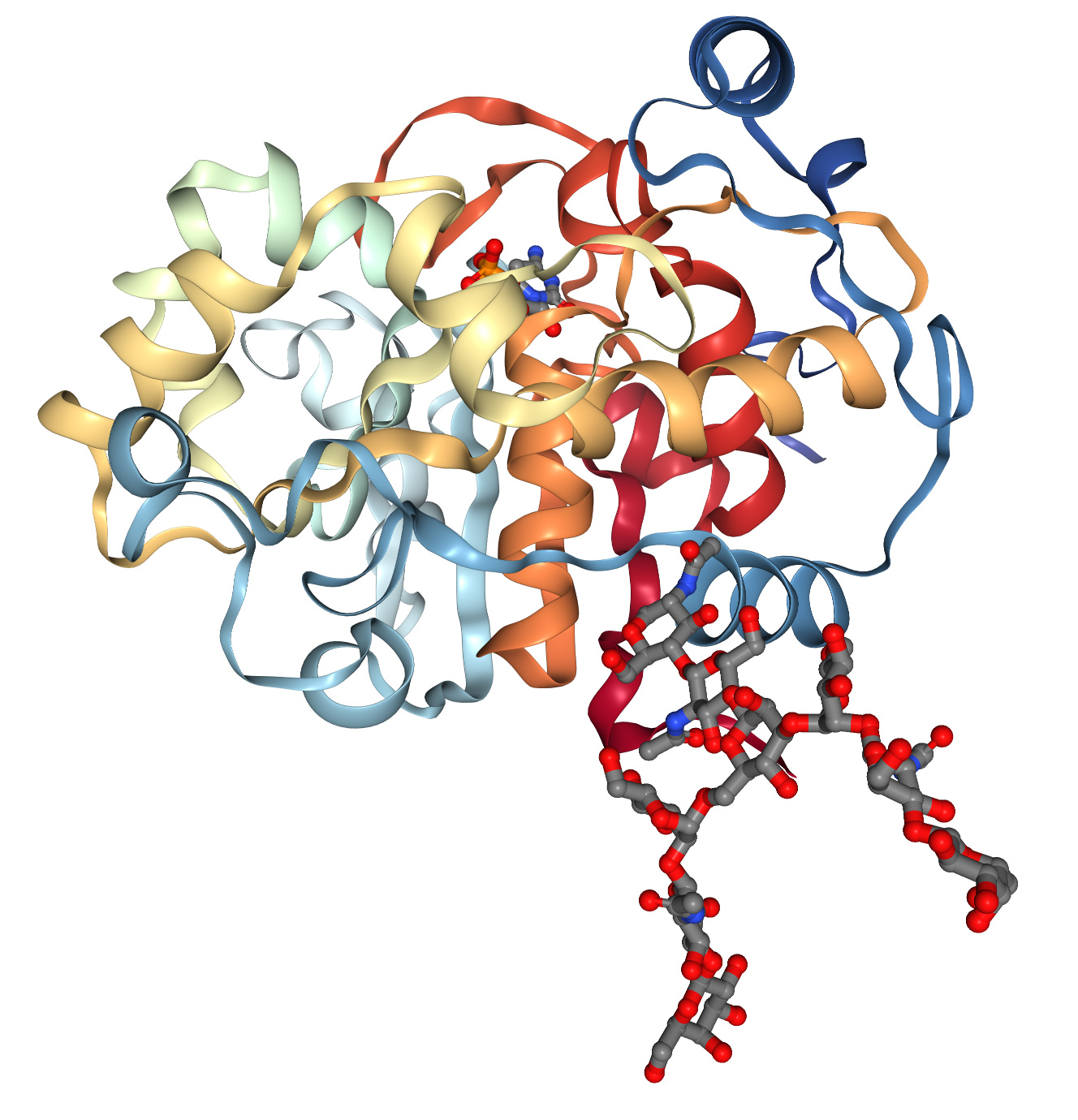
Predir les estructures de les proteïnes és difícil per diverses raons. En primer lloc, totes les possibles posicions tridimensionals de cada àtom en cada aminoàcid requereixen molta exploració. En segon lloc, les proteïnes aprofiten al màxim la complementarietat en la seva estructura química per configurar els àtoms de manera eficient. Com que les proteïnes solen tenir centenars de "donadors" d'enllaços d'hidrogen (normalment oxigen) que haurien d'estar a prop de l'"acceptor" d'enllaços d'hidrogen (normalment nitrogen unit a hidrogen), pot ser molt difícil trobar conformacions on gairebé tots els donants estiguin a prop de l'acceptor. En tercer lloc, hi ha exemples limitats per a l'entrenament de mètodes experimentals, per la qual cosa cal entendre les possibles interaccions tridimensionals entre aminoàcids sobre la base de seqüències unidimensionals utilitzant informació sobre l'evolució de les proteïnes rellevants.
La física es va utilitzar per primera vegada per modelar la interacció dels àtoms en la cerca de la millor conformació, i es va desenvolupar un mètode per predir l'estructura de les proteïnes. Karplus, Levitt i Warshel van rebre el Premi Nobel de Química 2013 pel seu treball sobre la simulació computacional de proteïnes. Tanmateix, els mètodes basats en la física són computacionalment cars i requereixen un processament aproximat, de manera que no es poden predir estructures tridimensionals precises. Un altre enfocament "basat en el coneixement" és utilitzar bases de dades d'estructures i seqüències conegudes per entrenar models mitjançant intel·ligència artificial i aprenentatge automàtic (IA-ML). Hassabis i Jumper apliquen elements tant de la física com de la IA-ML, però la innovació i el salt en el rendiment de l'enfocament provenen principalment de la IA-ML. Els dos investigadors van combinar creativament grans bases de dades públiques amb recursos informàtics de nivell industrial per crear AlphaFold.
Com sabem que han "resolt" el trencaclosques de la predicció estructural? El 1994 es va establir el concurs Critical Assessment of Structure Prediction (CASP), que es reuneix cada dos anys per fer un seguiment del progrés de la predicció estructural. Els investigadors compartiran la seqüència 1D de la proteïna l'estructura de la qual han resolt recentment, però els resultats de la qual encara no s'han publicat. El predictor prediu l'estructura tridimensional utilitzant aquesta seqüència 1D, i l'avaluador jutja independentment la qualitat dels resultats predits comparant-los amb l'estructura tridimensional proporcionada per l'experimentador (proporcionada només a l'avaluador). El CASP realitza revisions cegues reals i registra salts de rendiment periòdics associats amb la innovació metodològica. A la 14a Conferència CASP del 2020, els resultats de la predicció d'AlphaFold van mostrar un salt de rendiment tan gran que els organitzadors van anunciar que el problema de la predicció de l'estructura 3D s'havia resolt: la precisió de la majoria de les prediccions era propera a la de les mesures experimentals.
La importància més àmplia és que el treball de Hassabis i Jumper demostra de manera convincent com l'IA-ML podria transformar la ciència. La seva recerca mostra que l'IA-ML pot construir hipòtesis científiques complexes a partir de múltiples fonts de dades, que els mecanismes d'atenció (similars als de ChatGPT) poden descobrir dependències i correlacions clau en fonts de dades, i que l'IA-ML pot autojutjar la qualitat dels seus resultats. L'IA-ML és essencialment fer ciència.
Data de publicació: 23 de setembre de 2023